
The genome of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is about 80% similar to that of SARS-CoV. This high degree of similarity shared between these two viruses has led many to wonder why SARS-CoV-2 is so much more infectious and transmissible as compared to SARS-CoV.
A new Cells journal study compares the SARS-CoV-2 receptor-binding domain (RBD) (RBDCoV2) to the SARS-CoV RBD (RBDCoV) using computational methods to understand the binding affinities of the two proteins to the human receptor angiotensin-converting enzyme 2 (ACE2) receptor.
Study: Mechanistic Origin of Different Binding Affinities of SARS-CoV and SARS-CoV-2 Spike RBDs to Human ACE2. Image Credit: Cinefootage Visuals / Shutterstock.com
SARS-CoV-2 spike protein
Both SARS-CoV and SARS-CoV-2 utilize the ACE2 receptor for cell entry through their viral spike protein. The spike protein consists of three identical protomers that protrude from the lipid surface of the virus. Each protomer has two subunits known as S1 and S2. Whereas S1 is responsible for virus attachment to cells, S2 facilitates the fusion of the viral and cellular membranes.
The N-terminal domain (NTD) and C-terminal domain (CTD) of the S1 subunit fold independently as two large domains. The CTD acts as the RBD.
The SARS-CoV-2 RBD is the current target for coronavirus disease 2019 (COVID-19) messenger ribonucleic acid (mRNA) and adenovirus-based vaccines because it is majorly targeted by the immune response.
Spike protein dynamics
The spike protein trimer is not a rigid entity and instead presents different conformations or states. Moreover, this protein can present in a closed state, where all RBDs are in the down orientation and buried in the trimer. Comparatively, the spike protein can also present in an open state where one, two, or three RBDs are in the erect conformation. These orientations can coexist in equilibrium and be distributed within the spike population.
In the closed conformation, the spike protein cannot bind ACE2. A hinge-like motion progressively opens RBDs and allows ACE2 binding.
After binding of the first RBD to ACE2, the open conformation is stabilized and promotes the opening of the other two RBDs. These two RBDs now bind ACE2 in a fully open configuration, which further primes S2 unsheathing and results in membrane fusion.
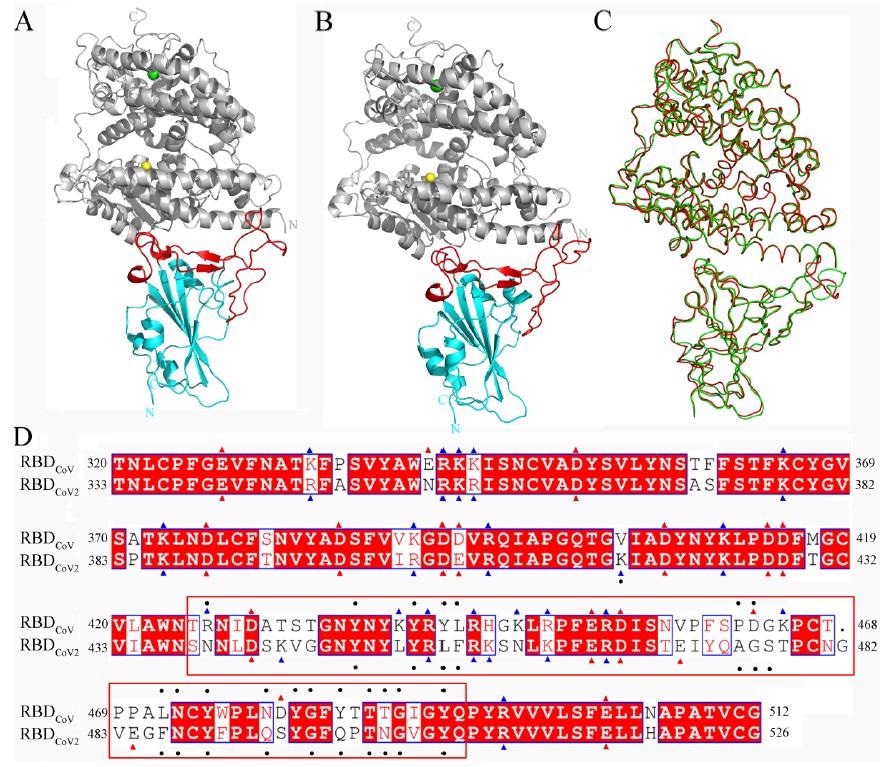
Structures of the RBDCoV-ACE2 and RBDCoV2-ACE2 complexes and sequence alignment of RBDCoV and RBDCoV2. (A,B) Cartoon representations of the complete complex structures of RBDCoV-ACE2 (modeled based on the crystal structure with PDB ID 2AJF and RBDCoV2-ACE2 (PDB ID: 6M0J respectively. ACE2 is colored gray, with Zn2+ and Cl− ions represented as spheres in yellow and green, respectively; cores and RBMs of both RBDs are colored cyan and red, respectively. (C) Backbone superposition of RBDCoV-ACE2 (red) and RBDCoV2-ACE2 (green). (D) Structure-based sequence alignment of RBDCoV and RBDCoV2. The identical residues are white on a red background and the similar residues are red on a white background; the negatively and positively charged residues are indicated by red and blue triangles, respectively. The ACE2-contacting residues (or RBD interface residues) identified in this work are indicated by black dots; RBM (residues 438–506 according to residue numbering of RBDCoV2) is highlighted by enclosure with a red box.
RBD structure and function
The opening of the RBD is a prerequisite for ACE2 binding. Even so, RBD is an independently folded domain and its opening has very little effect on the overall conformation.
Previous computational studies have shown that certain mutations outside the RBD can influence ACE2-binding affinity by altering the spike conformational dynamics. Yet, ACE2-binding affinity is usually evaluated using the RBD, rather than the spike trimer.
Several experimental and computational studies have shown that the ACE2-binding affinity of RBDCoV2 is higher than that of RBDCoV. Due to this greater binding affinity, SARS-CoV-2 has increased infectivity and transmissibility as compared to SARS-CoV.
RBDCoV and RBDCoV2 crystal structures in complex with human ACE2 reveal that the RBDs share similar overall conformations and nearly identical modes of ACE2 binding. Both RBDs have a core and a receptor-binding motif (RBM) subdomains.
The core has a twisted five-stranded antiparallel β-sheet that is connected by short helices and loops and contains few amino acids that encounter ACE2. The RBM has a short two-stranded antiparallel β-sheet, two short helices, and several long loops and contains most of the amino acids that make contact with ACE2.
RBDCoV and RBDCoV2 are 73.2% identical, whereas their cores are 88.0% identical and their RBMs are 47.8% identical. This may explain the different ACE2-binding affinities of RBDCoV and RBDCoV2, as the RBM has more ACE2-contacting amino acids.
About the study
The current study explores the mechanistic origin of the difference in the ACE2-binding affinities of RBDCoV and RBDCoV2. Molecular dynamics simulations were performed on the structures of RBD-ACE2 complexes of SARS-CoV and SARS-CoV-2.
Additionally, the researchers also conducted comparative dynamics and thermodynamics analyses, calculations of the protein-protein and per-residue binding free energies (BFEs), constructions of the interface residue contact networks (IRCNs), and comprehensive comparative analyses of IRCNs, interface interactions, and BFE components of individual amino acids.
Study findings
As compared to the RBDCoV2-ACE2 complex, RBDCoV-ACE2 demonstrates enhanced dynamics and inter-protein positional movements, as well as increased conformational entropy and conformational diversity. The inter-protein electrostatic attractive interactions primarily determine the high ACE2-binding affinities. Notably, the ACE2 and RBDCoV2 exhibit significantly enhanced electrostatic attractive interactions as compared to their interaction with RBDCoV.
The amino acid changes at the RBD interface are responsible for the overall stronger inter-protein electrostatic attractive force in RBDCoV2-ACE2. This tightens the interface packing and suppresses the dynamics of RBDCoV2-ACE2, as well as enhances the ACE2-binding affinity of RBDCoV2.
Since the RBD amino acid changes resulting in gain/loss of the positive/negative charges can greatly affect binding affinity, SARS-CoV-2 variants harboring such mutations warrant special attention, particularly those close to or at the binding interfaces of ACE2.
Conclusions
The current study provides new insights into the dynamics and energetics of the mechanisms of RBD-ACE2 interactions. Furthermore, the study findings explain an increased RBDCoV2-ACE2-binding affinity than that of RBDCoV.
- Zhang, Z., Xia, Y., Shen, J., et al. (2022) Mechanistic Origin of Different Binding Affinities of SARS-CoV and SARS-CoV-2 Spike RBDs to Human ACE2. Cells 11(8):1274. doi:10.3390/cells11081274.
Posted in: Molecular & Structural Biology | Medical Science News | Medical Research News | Disease/Infection News
Tags: ACE2, Adenovirus, Amino Acid, Angiotensin, Angiotensin-Converting Enzyme 2, binding affinity, Cell, Coronavirus, Coronavirus Disease COVID-19, covid-19, Enzyme, Genome, Immune Response, Membrane, Protein, Receptor, Respiratory, Ribonucleic Acid, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Spike Protein, Syndrome, Virus

Written by
Dr. Shital Sarah Ahaley
Dr. Shital Sarah Ahaley is a medical writer. She completed her Bachelor's and Master's degree in Microbiology at the University of Pune. She then completed her Ph.D. at the Indian Institute of Science, Bengaluru where she studied muscle development and muscle diseases. After her Ph.D., she worked at the Indian Institute of Science, Education, and Research, Pune as a post-doctoral fellow. She then acquired and executed an independent grant from the DBT-Wellcome Trust India Alliance as an Early Career Fellow. Her work focused on RNA binding proteins and Hedgehog signaling.
Source: Read Full Article