
Researchers in the United States have developed a new automated approach to detecting variants of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) – the agent that causes coronavirus disease 2019 (COVID-19) – that have increased growth rates over other lineages.
The repeated emergence of SARS-CoV-2 variants that exhibit increased transmissibility highlights the need for new approaches to detect and characterize new lineages rapidly, says Obermeyer and colleagues from the Broad Institute of MIT and Harvard in Cambridge, Massachusetts.
Now, the team has developed a multinomial logistic regression model called "PyR0" that can detect lineages of increasing prevalence.
By applying PyR0 to all publicly available SARS-CoV-2 genomes, the researchers pinpointed multiple mutations that increase transmissibility.
These included previously identified mutations in the viral spike protein, which mediates the initial stage of infection and many mutations within the nucleocapsid protein and non-structural proteins.
"PyR0 forecasts growth of new lineages from their mutational profile, identifies viral lineages of concern as they emerge, and prioritizes mutations of biological and public health concern for functional characterization," writes Obermeyer and the team.
A pre-print version of the research paper is available on the medRxiv* server, while the article undergoes peer review.
Repeated waves of SARS-CoV-2 have been driven by new, more transmissible variants
The COVID-19 pandemic has featured repeated waves of SARS-CoV-2 infection that have been driven by the emergence of new variants with increased transmissibility.
Rapidly identifying such viral lineages as they emerge and the ability to accurately forecast their dynamics is essential for guiding responses to outbreaks.
However, this effectively requires interrogation of the entire global SARS-CoV-2 genomic dataset, say Obermeyer and colleagues.
"The large size (currently over 2.5 million virus genomes) and geographic and temporal variability of the available data present significant challenges that will only become more acute as more viruses are sequenced," they write.
Furthermore, estimates of transmissibility that are based only on lineage frequency data do not harness the additional statistical power that can be obtained through analyzing the independent emergence and growth of the same mutation within multiple lineages.
"Performing a mutation-based analysis of lineage prevalence has the additional advantage of identifying specific genetic determinants of a lineage's phenotype, which is critically important both for predicting the phenotype of new lineages and for understanding the biology of transmission and pathogenesis," says the team.
What did the researchers do?
The team developed a hierarchical Bayesian regression model – PyR0 – that enables scalable analysis of all publicly available SARS-CoV-2 genomes.
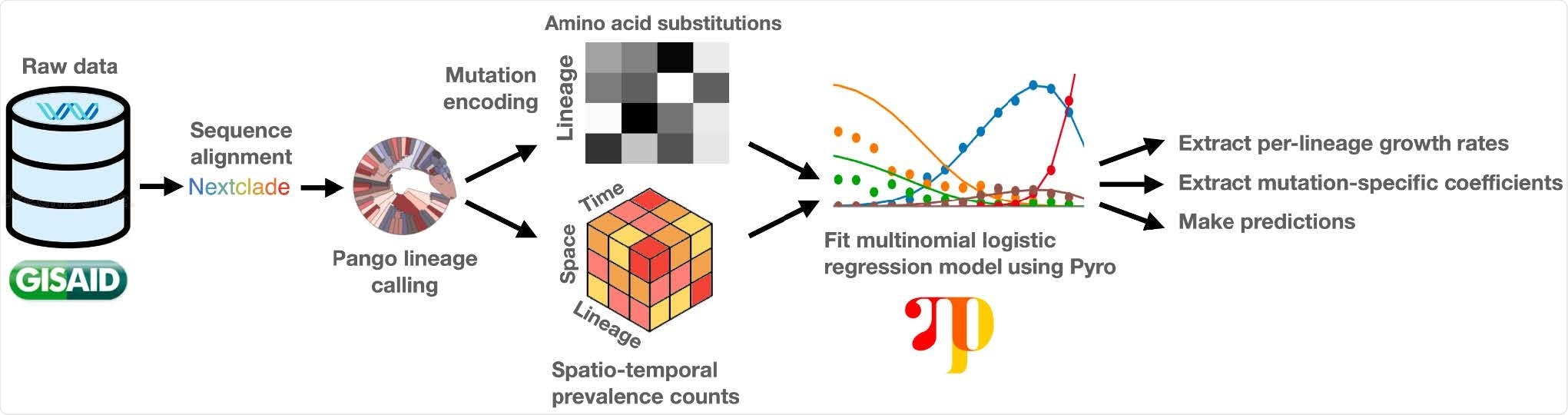
The model avoids the complexity of full phylogenetic inference by first clustering genomes based on their PANGO (Phylogenetic Assignment of Named Global Outbreak) lineages and then estimating the effect that each of the most common mutations within those lineages has on their growth rates.
By basing growth rate estimates on the contributions of individual mutations, PyR0 can be used to infer lineage growth rates, predict the growth rate of completely new lineages, forecast future lineage proportions, and estimate the effects of individual mutations on transmissibility, explains Obermeyer and colleagues.
The team applied PyR0 to all SARS-CoV-2 genomes (2,160,748) available on the Global Initiative On Sharing All Influenza Data (GISAID) platform as of July 6th, 2021, in a model that contained 1,281 PANGO lineages and 2,337 non-synonymous mutations.
What did they find?
The model's inferred growth rates exhibited a modest upward trend for all lineages and dramatically higher rates for several lineages that started to become more frequent in late 2020.
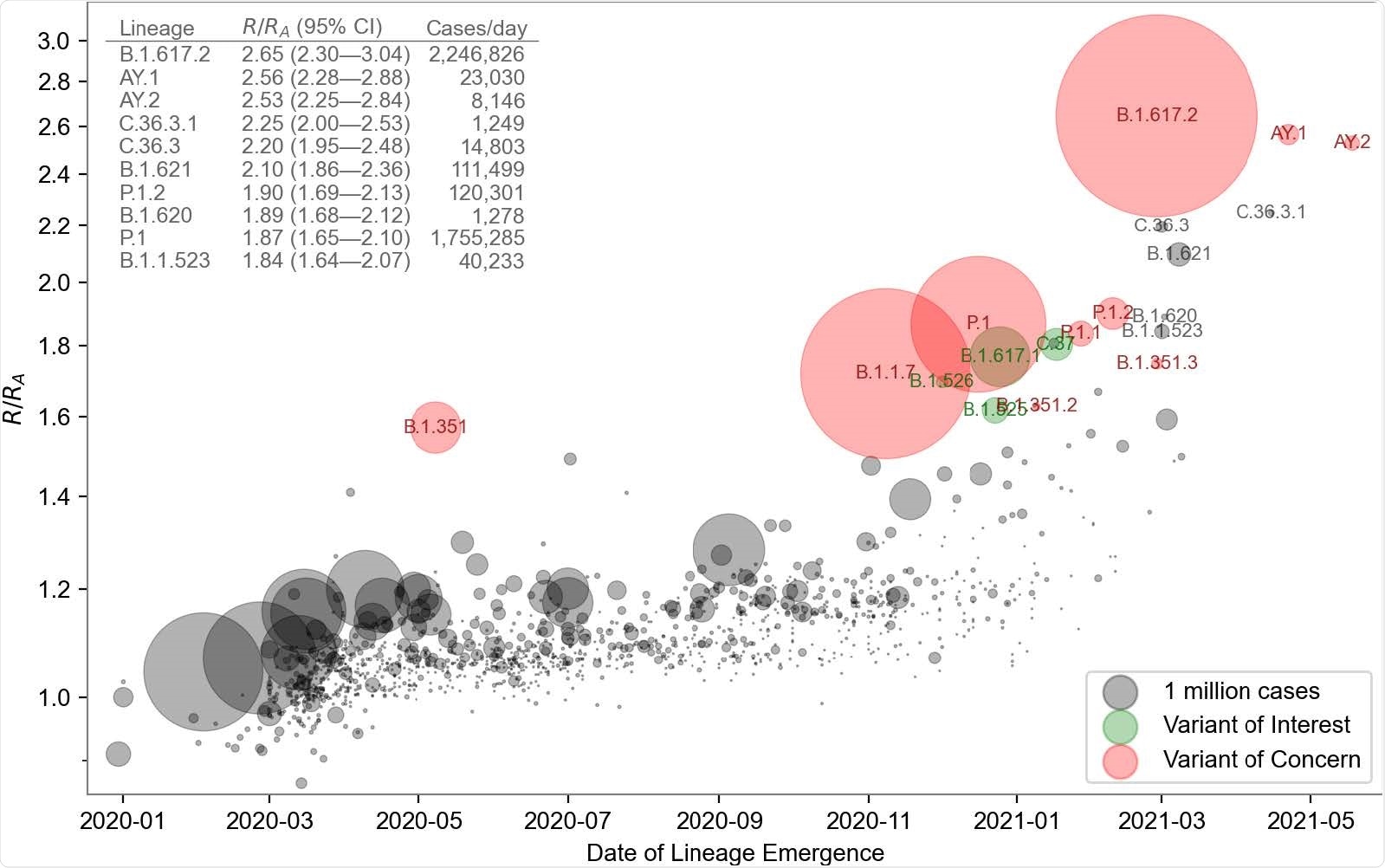
PyR0 correctly inferred that the B.1.617.2 (delta) variant has had the highest growth rate to date and predicted that this variant and its sublineages would displace other lineages, including the previously dominant B.1.1.7 (alpha) variant.
Obermeyer and colleagues say the model would have provided early warning of an increase in variants of concern if it had been routinely applied to available SARS-CoV-2 data. For instance, PyR0 would have predicted the oncoming dominance of B.1.1.7 in early November 2020, whereas the first models predicting this were published in January 2021.
"A similar prediction would have been available for B.1.617.2 by late April 2021," they add.
PyR0 identified numerous important spike and non-spike mutations
The PyR0 model identified multiple substitutions in functional regions of the SARS-CoV-2 spike protein that are associated with increased transmissibility, including D614G, L452R, and ΔH69V70.
Another cluster of growth rate-enhancing mutations was identified at positions 160–210 of the nucleocapsid protein.
"Although previously uncharacterized, mutations in this region were recently linked to increased efficiency of SARS-CoV-2 RNA packaging," say the researchers.
The highest concentration of growth rate-associated mutations with predictive power was found in the non-structural proteins (nsp) 2, 4, 6, and nsp 12–14, which the researchers say points to unexplored function at those sites:
"For example, nsp4 and nsp6 have roles in assembly of replication compartments, and substitutions in these regions may influence the kinetics of replication."
What did the authors conclude?
Obermeyer and colleagues say that once applied to the full set of publicly available SARS-CoV-2 genomes, PyR0 can be used to analyze mutations that drive increased transmissibility, identify experimentally established driver mutations in spike and highlight the role of non-spike mutations.
"The highlighted genetic diversity offers promising targets for follow-up investigation and may open new avenues for therapeutic or public health intervention," they conclude.
*Important Notice
medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Obermeyer FH, et al. Analysis of 2.1 million SARS-CoV-2 genomes identifies mutations associated with transmissibility. medRxiv, 2021. doi: https://doi.org/10.1101/2021.09.07.21263228, https://www.medrxiv.org/content/10.1101/2021.09.07.21263228v1
Posted in: Medical Science News | Medical Research News | Disease/Infection News
Tags: Amino Acid, Coronavirus, Coronavirus Disease COVID-19, Frequency, Genetic, Genomic, Influenza, Mutation, Pandemic, Phenotype, Protein, Public Health, Research, Respiratory, RNA, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Spike Protein, Syndrome, Virus

Written by
Sally Robertson
Sally first developed an interest in medical communications when she took on the role of Journal Development Editor for BioMed Central (BMC), after having graduated with a degree in biomedical science from Greenwich University.
Source: Read Full Article