. DOI: 10.1016/j.ccell.2023.06.010")
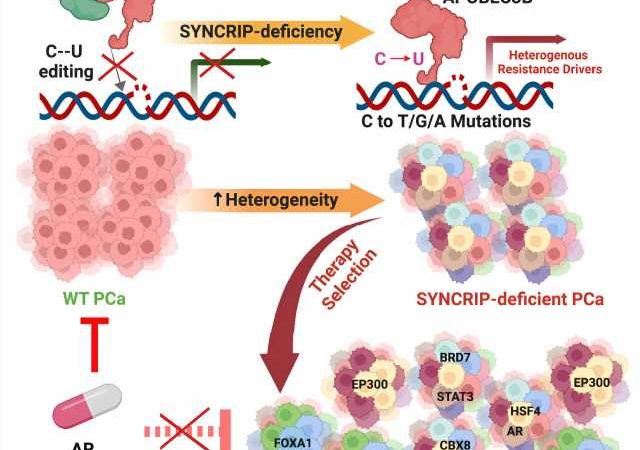
Loss of a gene known as SYNCRIP in prostate cancer tumors unleashes cellular machinery that creates random mutations throughout the genome that drive resistance to targeted treatments, a team led by UT Southwestern Medical Center researchers has discovered. The findings, published in Cancer Cell, could lead to new interventions that thwart this process in prostate and other cancer types, making them far easier to treat.
“This study paves the way for innovative strategies to fix the broken ‘brake’ for mutagenesis in cancer and curb resistance to treatments,” said study leader Ping Mu, Ph.D., Assistant Professor of Molecular Biology at UT Southwestern and a member of the Harold C. Simmons Comprehensive Cancer Center.
Researchers have long known that many cancers are typically initiated by mutations that develop in DNA and that additional mutations frequently form, driving tumors to grow and spread. Although drugs exist to target tumors bearing specific mutations, they don’t cover the gamut of mutations that arise in cancer cells, Dr. Mu explained. Thus, the more mutations present in a tumor, the more resistant it becomes to treatment. But how and why these additional mutations come about so quickly have been unknown.
To answer these questions, Dr. Mu and his colleagues looked for genetic differences in the cells of prostate cancer tumors—the focus of the Mu lab—from samples taken from xenografted human tumors before and after the administration of antiandrogen therapy, a common treatment that starves prostate cancers of sex hormones that can fuel their growth. Although this treatment is often initially effective, prostate tumors nearly always become resistant to antiandrogen therapy over time.
In about half of the post-treatment samples, the SYNCRIP gene had disappeared. The researchers also found a variety of mutations that bore the signature of APOBEC proteins, cellular machinery that produces a specific type of mutation used for immune defense in healthy cells.
Further experiments suggested that SYNCRIP serves as a brake for a major member of the APOBEC protein family, known as APOBEC3B, keeping its activity in check. When SYNCRIP is lost, Dr. Mu explained, APOBEC3B’s function becomes uncontrolled, causing it to create random mutations throughout the genome. When the researchers genetically altered cells to remove APOBEC3B, they didn’t accumulate mutations, even in the absence of SYNCRIP, confirming that their roles are linked.
Searching for genetic drivers of treatment resistance that arise from this process, the researchers scanned the genomes of post-treatment cells for APOBEC-caused mutations that prompted genes to become overactive. They identified eight genes that appear to be hot spots for APOBEC-driven mutations, all of which made antiandrogen therapy ineffective. When the researchers deleted these genes from the cells, they became responsive to therapy again.
Because mutations with APOBEC signatures are found in about 70% of human cancers, Dr. Mu said, the findings shed light on several mysteries in oncology. Those include why cancerous tumors tend to develop so many mutations quickly and why the mutation pattern is heterogeneous for different patients with the same cancer type and often among different cells in the same tumor.
Currently, cancers are treated by addressing these mutations individually, he added, which is effective only if a targeted treatment exists for a specific mutation. In the future, researchers may be able to develop treatments aimed at restoring SYNCRIP or inhibiting APOBEC proteins in order to prevent resistance-driving mutations before they develop.
Dr. Mu is a Deborah and W.A. “Tex” Moncrief, Jr. Scholar in Medical Research.
More information:
Xiaoling Li et al, Loss of SYNCRIP unleashes APOBEC-driven mutagenesis, tumor heterogeneity, and AR-targeted therapy resistance in prostate cancer, Cancer Cell (2023). DOI: 10.1016/j.ccell.2023.06.010
Journal information:
Cancer Cell
Source: Read Full Article